What needs to be reported to the IRB
 Natalie Ross
Natalie Ross An Adverse Event must be reported to the IRB if it: (i) is more likely than not related to study activities; and (ii) represents a new risk; and (iii) is unanticipated. In addition, an expected event that is occurring at a frequency or intensity greater than originally anticipated must be reported to the IRB.
What should be reported to the IRB?
Investigators are required to report promptly “to the IRB… all unanticipated problems involving risks to human subjects or others,” including adverse events that should be considered unanticipated problems (21 CFR 56.108[b][1], 21 CFR 312.53[c][1][vii], and 21 CFR 312.66).
What is considered a reportable event?
A reportable event is any event that the IRB may determine is an unanticipated problem involving risks to subjects or others or serious or continuing noncompliance with the federal regulations or Institutional Review Board requirements.
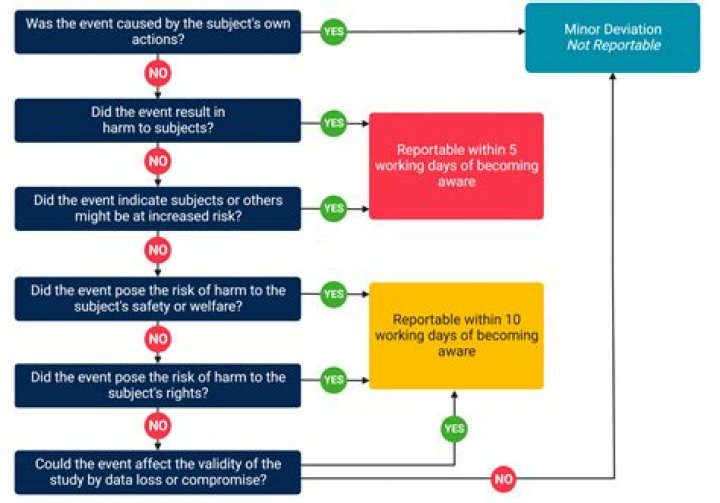
What types of events must be reported to the IRB within 5 business days of becoming aware of the event?
Events that result in harm to subject’s rights, safety or welfare must be reported to the IRB within 5 business days of knowledge of the event. Events that result in harm to the integrity of the data must be reported to the IRB within 10 business days of knowledge of the event. 2.Do all SAEs need to be reported to the IRB?
All SAEs must be reported to the IRB within 5 business days as “reportable new information.”
What are the main criteria for serious adverse event?
Report if the adverse event resulted in a substantial disruption of a person’s ability to conduct normal life functions, i.e., the adverse event resulted in a significant, persistent or permanent change, impairment, damage or disruption in the patient’s body function/structure, physical activities and/or quality of …
Do all adverse events need to be reported?
The FDA does not receive reports for every adverse event or medication error that occurs with a product. Many factors can influence whether an event will be reported, such as the time a product has been marketed and publicity about an event.
Do you need IRB approval for cadaver research?
Research Involving Deceased Persons or Cadavers If the data or samples contain personal identifiers, the research is subject to HIPAA. … If the information collected from a cadaver will result in an investigator obtaining information about the cadaver’s living relatives (e.g., genetic studies) IRB approval is required.What types of study events require immediate IRB review?
Federal regulations and IRB policy require study teams to submit reportable events to the IRB for review. Reportable events include noncompliance, new information, serious adverse event, and unanticipated problems.
What information should be provided to an IRB for review at the initiation of a study?The study protocol (and amendments), the information to be given to the subject (informed consent, advertisements), the Investigator Brochure (or drug label), any other relevant safety information, and an outline of the qualifications of the investigator.
Article first time published onWhat does reportable mean?
Definition of reportable 1 : worth reporting reportable news. 2 : required by law to be reported reportable income reportable diseases.
What are considered serious reportable incidents?
A serious reportable event (SRE) is an incident involving death or serious harm to a patient resulting from a lapse or error in a healthcare facility.
Are Never events reportable?
Since the NQF disseminated its original Never Events list in 2002, 11 states have mandated reporting of these incidents whenever they occur, and an additional 16 states mandate reporting of serious adverse events (including many of the NQF Never Events).
What is an IND safety report?
The phrase “IND safety reports” originates in FDA regulations 21 CFR 312 – Investigational New Drug Application. … The IND safety reports concern a product under study and such reports may not necessarily apply to events that occurred in the protocol conducted at JHM.
Is this an example of an unanticipated problem that requires reporting to the IRB?
Is this an example of an unanticipated problem that requires reporting to the IRB? No, this does not need to be reported because it was assessed by the researcher as unrelated to the research study. A researcher conducts a focus group to learn about attitudes towards hygiene and disease prevention.
Is death an SAE?
A Serious Adverse Event (SAE) is defined by FDA and NCI as any adverse drug event (experience) occurring at any dose that in the opinion of either the investigator or sponsor results in any of the following outcomes: death, a life threatening adverse drug experience, inpatient hospitalization or prolongation of …
What is the required reporting timeline for reporting patient safety events?
The California Department of Public Health (CDPH) and Medi-Cal both mandate the reporting of events within 5 Days of the event’s discovery.
Which of the following qualify for reporting as an adverse event or a special situation?
Adverse event can therefore be: any adverse or unintended sign (for example, an abnormal laboratory finding) symptom. disease temporarily associated with the use of a medicinal product, whether ornot a causal relationship with the treatment)”
What do you report in pharmacovigilance?
- Individual Case Safety Report (ICSR)
- Case medical information inquiries.
- Product complaints.
- Reports from medical representatives.
- Reports from competent authorities.
- Contractual partners (co-marketed products, in-licensing, out-licensing, and distribution partners).
How do you report serious adverse events?
- Report Online.
- Consumer Reporting Form FDA 3500B. Follow the instructions on the form to either fax or mail it in for submission. …
- Call FDA at 1-800-FDA-1088 to report by telephone.
- Reporting Form FDA 3500 commonly used by health professionals. View Instructions for Form FDA 3500.
What are the 3 common factors of an adverse event?
The most common con- tributing factors were (i) lack of competence, (ii) incomplete or lack of documenta- tion, (iii) teamwork failure and (iv) inadequate communication. Conclusions: The contributing factors frequently interacted yet they varied between different groups of serious adverse events.
When Should serious adverse events be reported?
An investigator (if he/she is not a sponsor-investigator) must report to the sponsor any serious adverse event within 24 hours of investigator learning about the event, whether or not considered drug related, including those listed in the protocol or investigator brochure and must include an assessment of whether there …
Who reporting adverse events in clinical trials?
In clinical research, a researcher-doctor must report any adverse event to the ethics committee, institution, the office of DCGI and the sponsor (if any) and manage the adverse event without imposing any financial burden to the research participant.
Who is responsible for reporting directly to FDA?
Manufacturers: Manufacturers are required to report to the FDA when they learn that any of their devices may have caused or contributed to a death or serious injury. (Key terms are defined in 21 CFR 803.3.) Instructions are available for completing the required 3500A form.
Who can report an adverse event?
Reporting of adverse events from the point of care is voluntary. FDA receives some adverse event and medication error reports directly from health care professionals (such as physicians, pharmacists, nurses and others) and consumers (such as patients, family members, lawyers and others).
What research does not require IRB approval?
Publicly available data do not require IRB review. Examples: census data, labor statistics. Note: Investigators should contact the IRB if they are uncertain as to whether the data qualifies as “publicly available.”
What types of research require IRB approval?
FDA regulations generally require IRB review and approval of research involving FDA-regulated products (e.g., investigational drugs, biological products, medical devices and dietary supplements) (21 CFR Part 56).
Do anonymous surveys need IRB approval?
Do anonymous surveys (with no way to connect data with subjects) need IRB review? Answer • Yes, but anonymous surveys qualify as exempt. You still need to submit to IRB, which determines if project is exempt.
Which of the following materials must be included in an IRB application?
- Application.
- Consent Document(s)
- Recruitment Materials.
- Study Instrument(s)
- Permission Letters (if applicable)
- Certificate of Education (if not already on file)
- Grant proposal narrative (if applicable)
- The application materials have been made into one PDF.
What are the 4 important ethical issues IRB guidelines address?
- Respect for persons: respect for patient autonomy.
- Beneficence: maximize benefits and minimize harm.
- Justice: Equitable distribution of research burdens and benefits.
What are the minimum elements that are typically required for an IRB protocol?
Common Protocol Elements At a minimum, IRB protocols should contain the Objectives, Methods, Quality Control and Assurance, Ethics/Protection of Human Subjects, and Data Handling and Record Keeping.